![]()
Computational Structural Biology: SAXS
Supplementary Figures and Data
These figures supplement
the manuscript "Using Situs for the Registration of Protein Structures
with Low-Resolution Bead Models from X-ray Solution Scattering", by Willy
Wriggers and Pablo Chacón, Journal of Applied Crystallography 34:773-776, 2001.
![]()
(A) Mapping a High-Resolution Structure to a Hexagonal Close-Packed (HCP) Lattice.
As described in the manuscript, we mapped the atomic structures of ten trial proteins to HCP lattices with bead radii from 1 to a maximum of 20 Angstrom. The modeling was stopped for radii that would result in less than three beads. The resulting models served as "simulated" low-resolution data to test the performance of our docking algorithm. Shown here is the example of ovalbumin: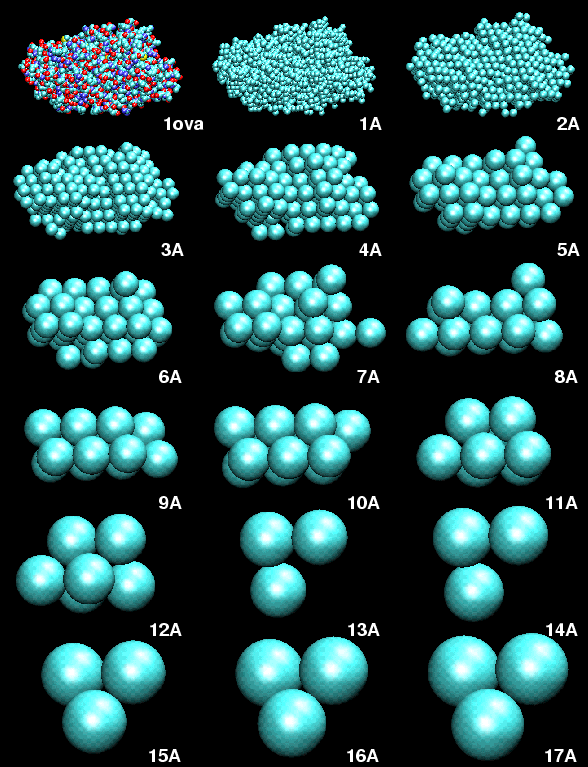
(Click image to enlarge)
Figure 1. HCP bead models of ovalbumin (PDB entry 1ova). Shown are the initial structure in van-der-Waals sphere representation, and the generated HCP bead models, as a function of bead radius in Angstrom.
![]()
(B) Precision of the Docking
The results of the docking of the ten trial proteins are characterized by their deviation from the original position:A.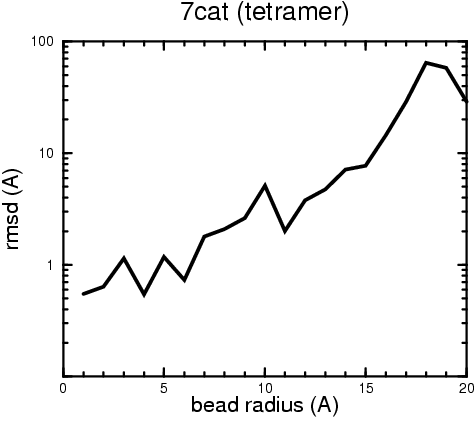 B.
B.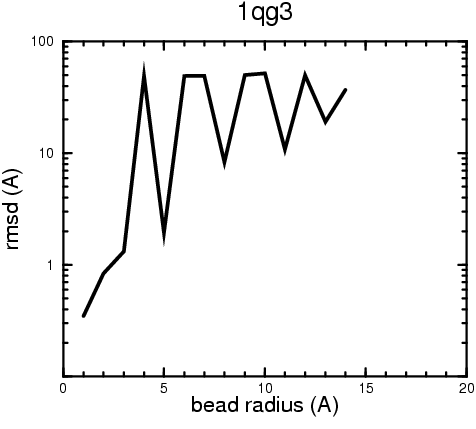 C.
C. D.
D.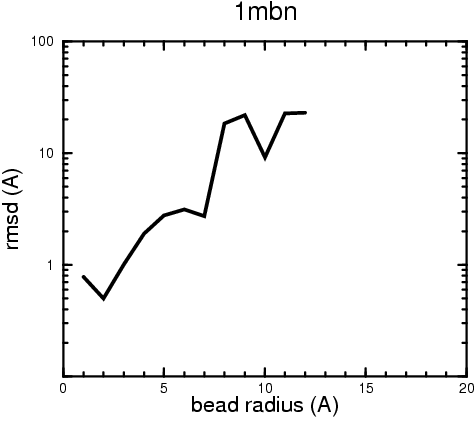 E.
E.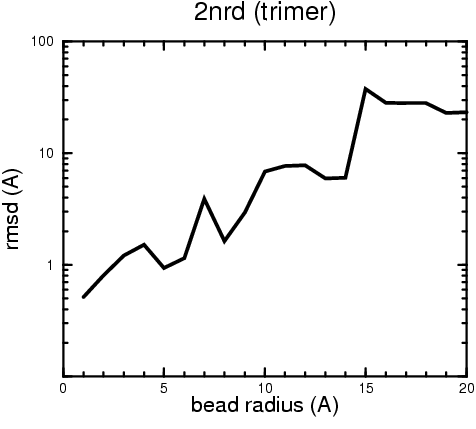
F.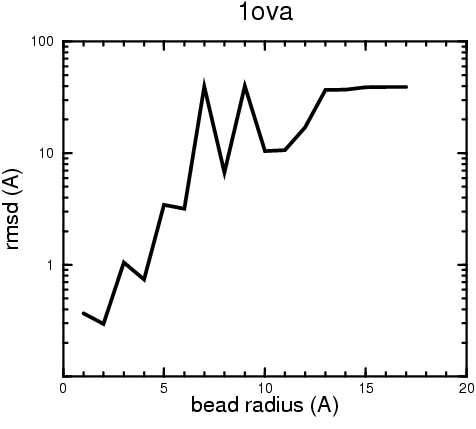 G.
G.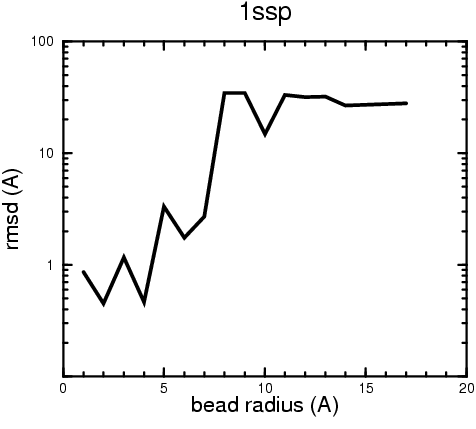 H.
H.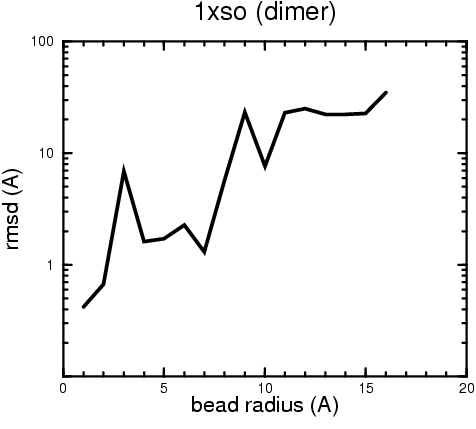 I.
I. J.
J.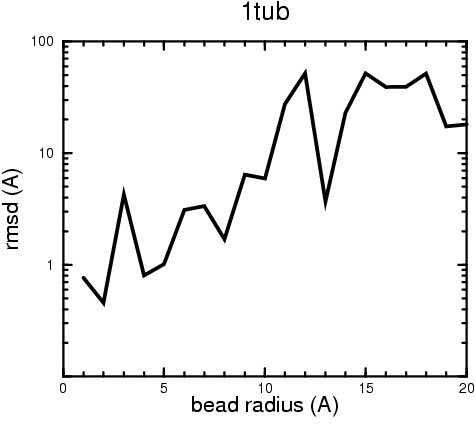
(Click thumbnails to enlarge)
Figure 2. Root-mean-square deviation (rmsd) of the atomic structures, after they have been fitted to the bead models using Situs, from the same structures in the initial position. The rmsd is shown as a function of bead radius. A: catalase (PDB entry 7cat); B: beta-4-integrin (1qg3); C: chymotrypsinogen A (2cga); D: myoglobin (1mbn); E: nitrito-reductase (2nrd); F: ovalbumin (1ova); G: spermadhesin PSPI/PSPII (1spp); H: superoxide dismutase (1xso); I: troponin C (1top); J: alpha-beta tubulin (1tub). The cases correspond to unique matches, except E (6-fold degeneracy), A (4-fold degeneracy), and C,D,H (2-fold degeneracy). In the A,C,D,E,H cases only the best fit among the degenerate highest scoring solutions was evaluated for the Figure.
![]()
(C) Docking of Atomic Structures to Experimental SAXS Bead Models
Following the steps described in the SAXS docking tutorial, we have brought the atomic structures of the ten trial proteins into register with actual SAXS bead models from the Andreu lab in Madrid (Chacón et al., J. Mol. Biol. (2000) 299:1289-1302):A. B.
B.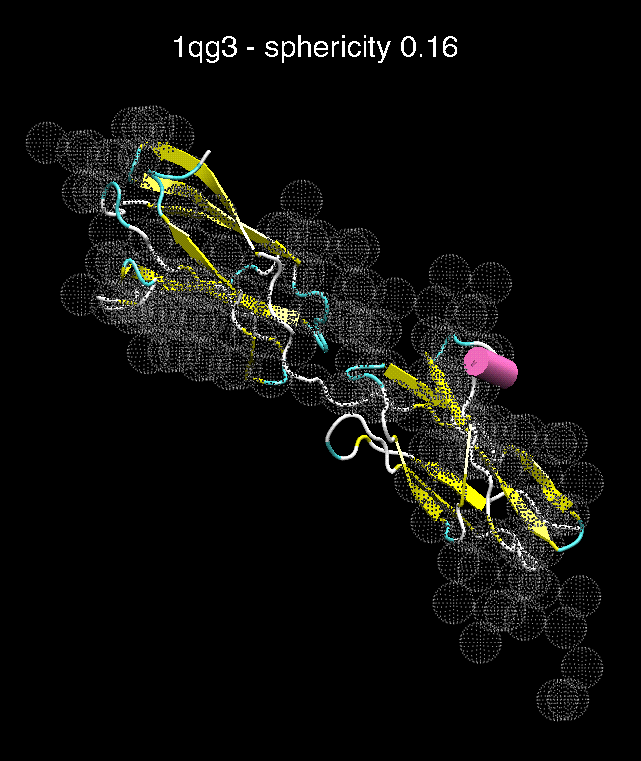 C.
C. D.
D.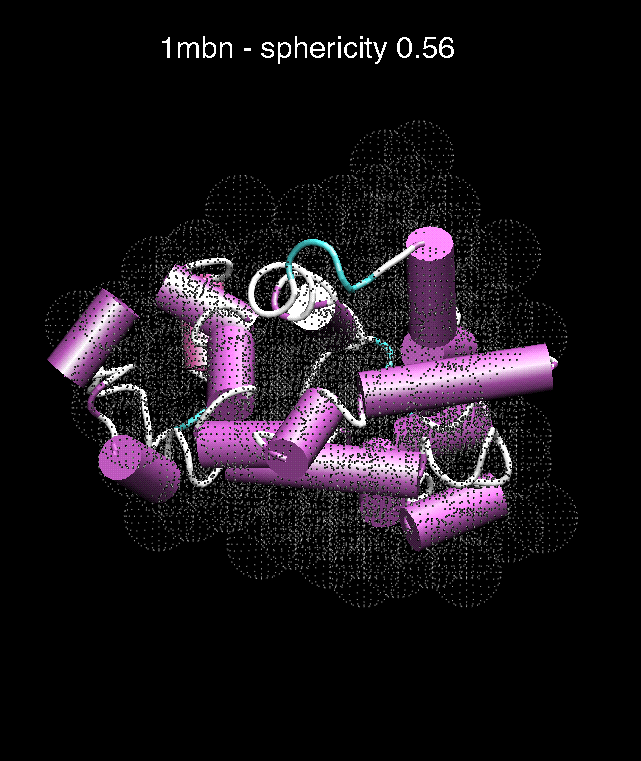 E.
E.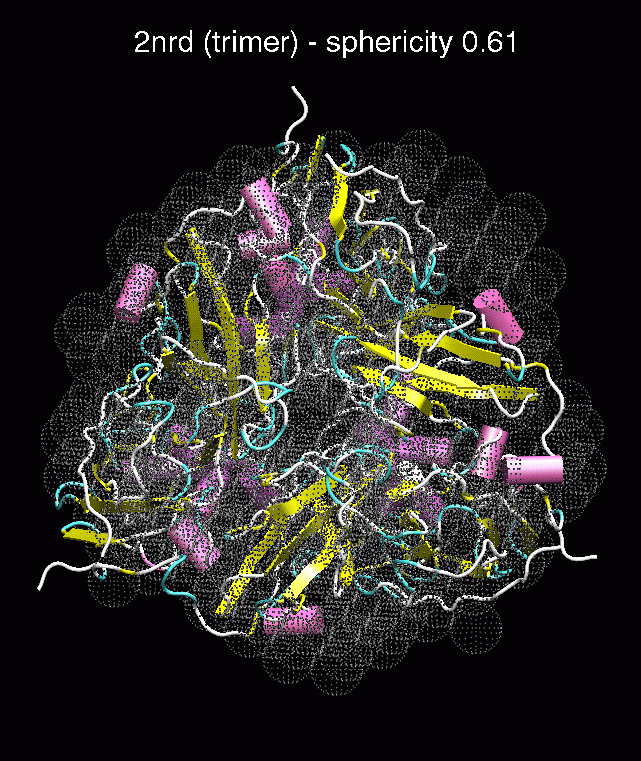
F.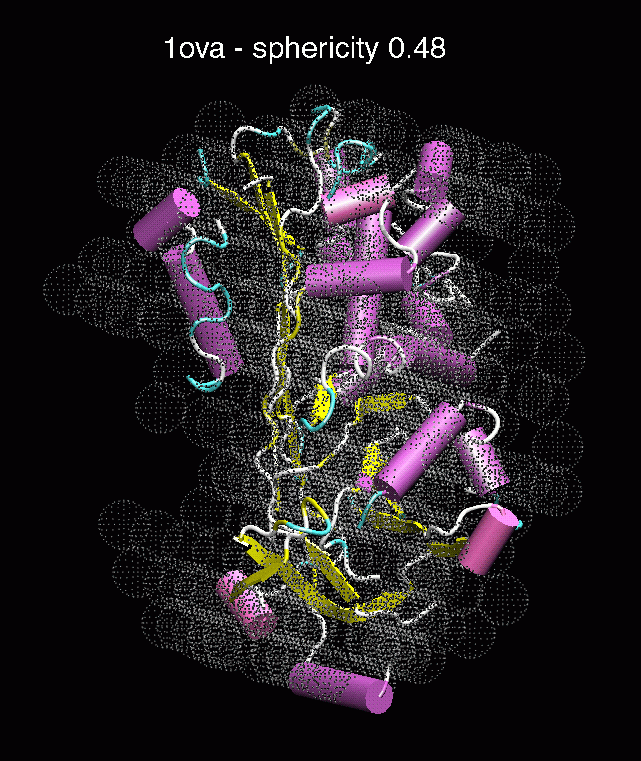 G.
G.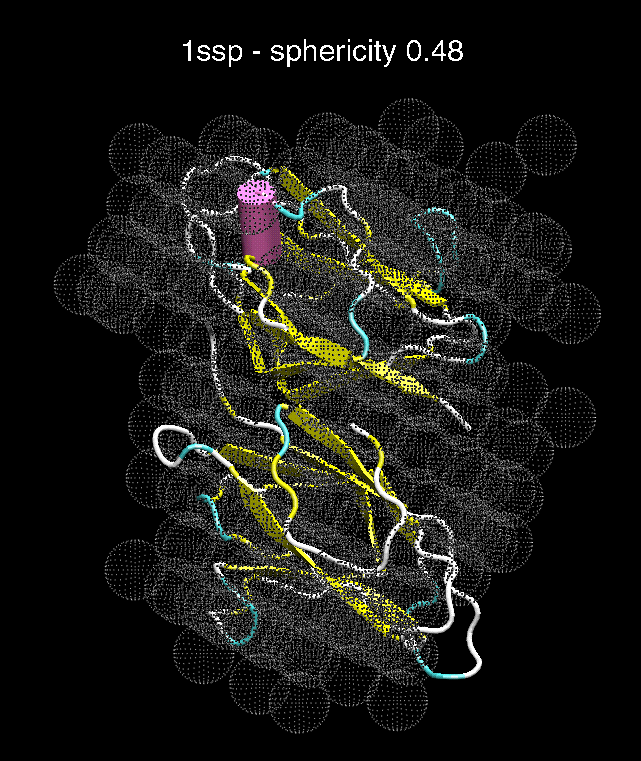 H.
H.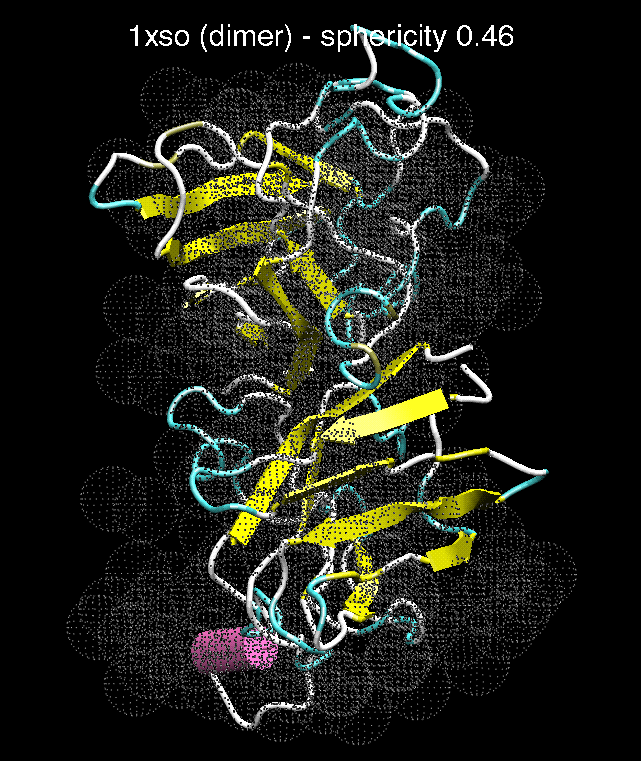 I.
I. J.
J.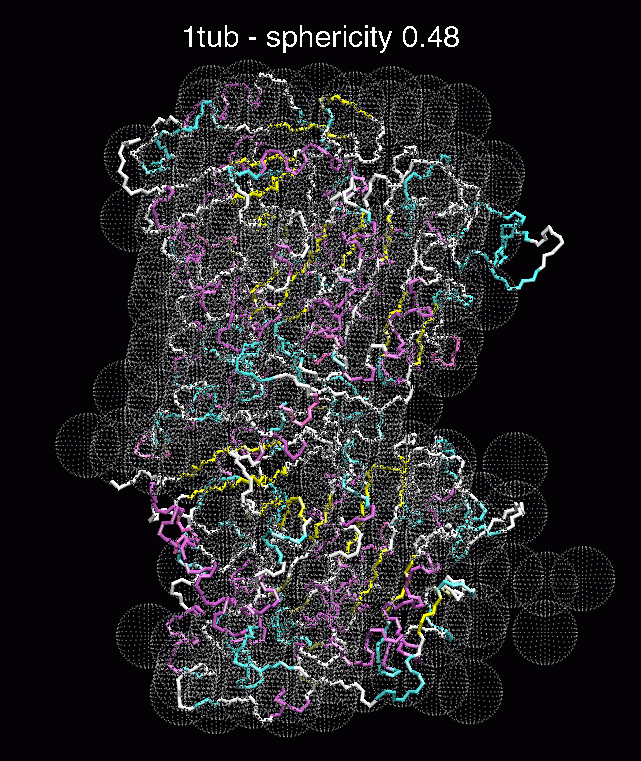
(Click thumbnails to enlarge)
Figure 3. The optimum matches of ten trial proteins with SAXS bead models, generated using Situs. Also shown is the sphericity of the atomic structures (for a definition see the manuscript). A: catalase (PDB entry 7cat); B: beta-4-integrin (1qg3); C: chymotrypsinogen A (2cga); D: myoglobin (1mbn); E: nitrito-reductase (2nrd); F: ovalbumin (1ova); G: spermadhesin PSPI/PSPII (1spp); H: superoxide dismutase (1xso); I: troponin C (1top); J: alpha-beta tubulin (1tub).
![]()
Useful Links and Further Information
![]() Situs SAXS Tutorial
Situs SAXS Tutorial
![]() Home Page of the Corresponding Author
Home Page of the Corresponding Author
![]()